Clinical Trials Are Built Around Everyone But the Patient
Late last year, my long-time friend Brian Blackmon came to me with a problem that started with his father. His dad had been enrolled in a clinical trial and, like most participants, was handed a thick stack of documents at the consent appointment and largely left to figure it out on his own. No digital tools. No plain-language reference materials. No easy way to look up what he was supposed to do, or why, or what to expect next.
Brian had spent his career in clinical research. He knew this wasn't unusual. What struck him was the contrast: every other stakeholder in a clinical trial has purpose-built digital tools. Sponsors have trial management systems. Sites have CTMS platforms. Coordinators have eSource and EDC tools. Participants have a PDF and a phone number.
That asymmetry is what became Clear Trials. And over the past five months, it's shaped how I think about what the clinical trial industry has been quietly getting wrong.
The experience gap
The clinical trial industry invests enormous resources in recruitment — getting participants enrolled. It invests comparatively little in what happens after enrollment, which is where participants actually live out the trial.
A 2024 study published in The Lancet's eClinicalMedicine found that across 798 federally funded trials, the average consent form was written at a 12th grade reading level — significantly above the 8th grade average reading level of U.S. adults. Each additional grade level increase in a consent form was associated with a 16% higher dropout rate. Dropout is one of the most persistent and costly problems in clinical research. Trials that lose participants lose data quality, timeline, and in some cases, viability. And the evidence increasingly points to comprehension as a root cause.
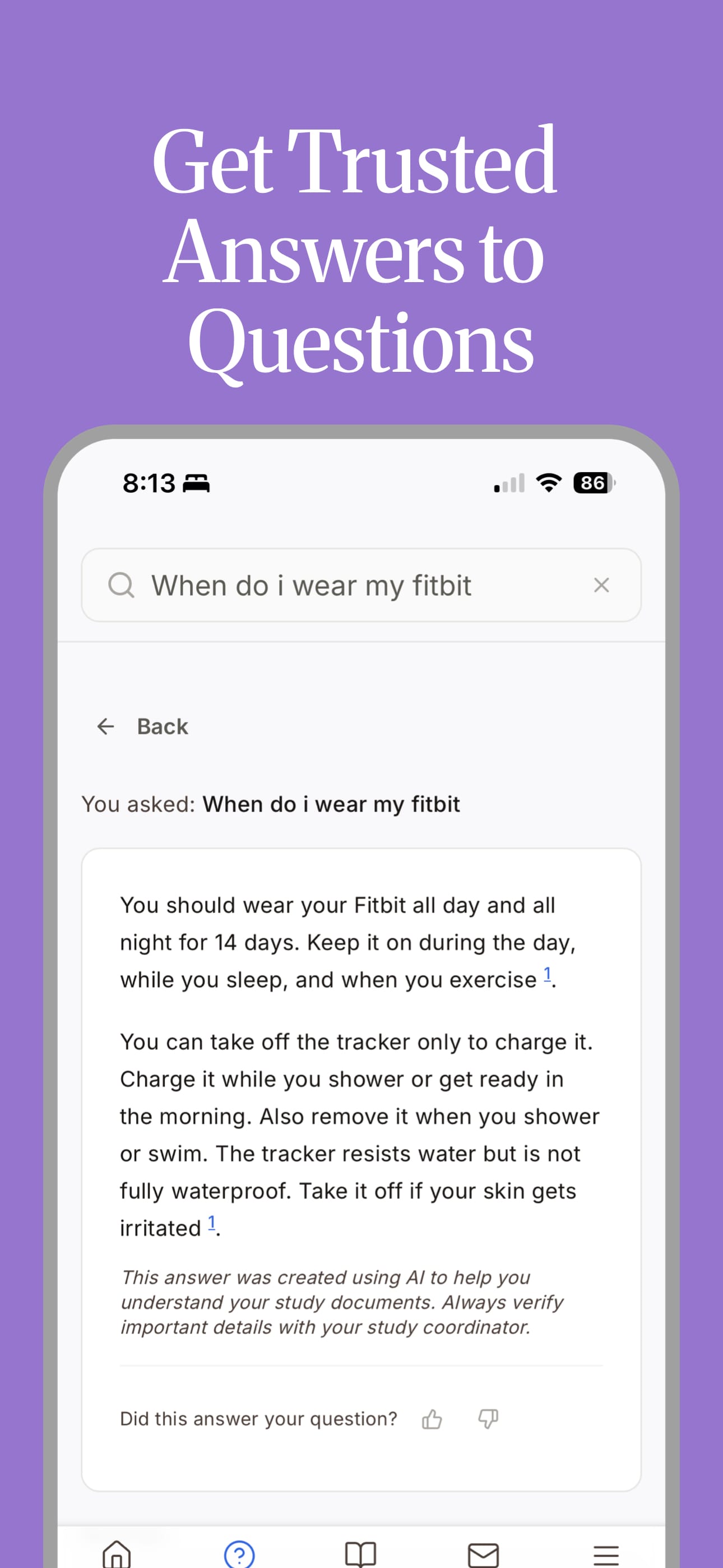
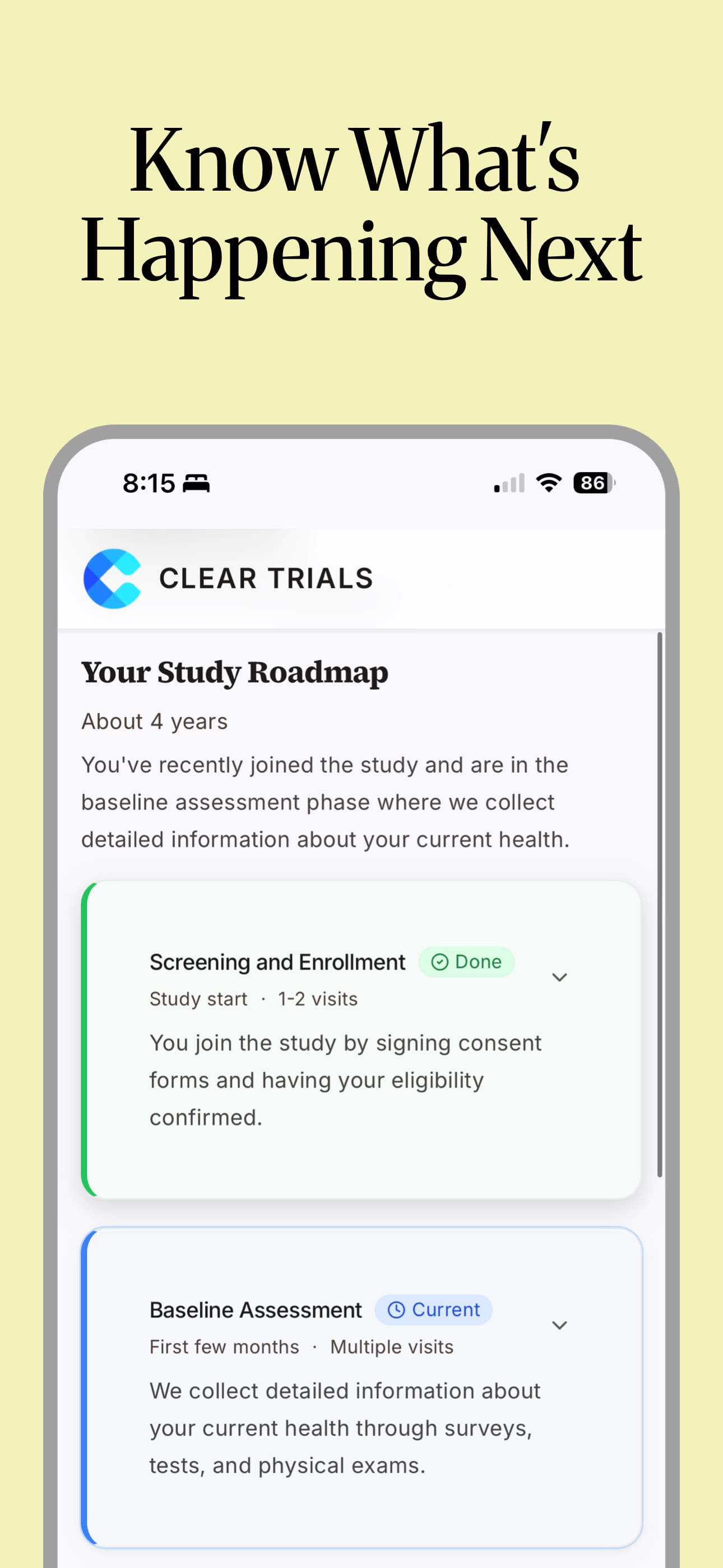
Consent documents can run 30, 40, sometimes 60 pages. They're carefully written and IRB-approved. They're also written for regulators, not for people. Once participants go home, they're expected to manage complex medical protocols — visit schedules, medication restrictions, home procedure instructions, reporting requirements — sometimes for months or years, with documents that were never really designed to help them do that.
When they have questions, they call their coordinator who is managing dozens of other participants across multiple trials, already stretched thin. When they don't understand something and don't call, they make their best guess. And that shows up downstream as protocol deviations, monitoring findings, no-shows, and eventually, dropout.
The industry frames this as a retention problem. I think it's something more basic: participants have never been treated as the primary user of their own trial experience. The tools have been built for everyone else.
What we built — and what we didn't
Clear Trials takes IRB-approved trial documents and transforms them into plain-language, accessible content that participants and their caregivers can actually use throughout a trial. Not a replacement for consent. Not a recruitment platform. Not a chatbot generating its own clinical claims.
The source documents stay the source of truth. We built tools to make them usable for the people who actually need to use them.
We also built caregiver access from the beginning because in many cases, the person managing a participant's trial experience isn't the participant. It's a spouse, a parent, an adult child. If they don't understand the trial, neither does the participant in any practical sense. The industry talks a lot about participant experience. It talks almost nothing about the family member driving them to appointments and helping them remember their prep instructions.
Coordinators are a real part of this too. We designed the platform so it reduces their communication burden — fewer calls about basic protocol questions — while giving them better visibility into where participants might be struggling. They're not the primary user. But this only works if it makes their job easier.
The thing that surprised me most
I came in expecting regulatory requirements to be the hardest constraint to design around. They turned out to be among the most useful things we have.
Because trial documents are IRB-approved, there's a clear, reliable source of truth. We're not generating content from scratch or making clinical claims. We're improving access to what already exists — translating documents designed for compliance into materials designed for comprehension. That distinction matters in a space built on trust, and it creates a defensible position that a lot of AI health tools don't have. It took me a few months to stop seeing the regulatory environment as a limitation and start seeing it as a foundation.
The second thing: the moat isn't AI. Every healthtech company is building AI features right now. What actually matters in clinical trials is trust, compliance, and relationships with the sites and sponsors who have to bet their trial data quality on your platform. The technology serves those things. It's not the headline.
Where we are and where this goes
We're currently in conversations with CROs and site networks who see the same gap we do. The immediate focus is identifying the right trials to pilot the platform — proving its value in a real clinical trial setting and learning what we don't yet know. Brian has first-hand experience doing this from inside Duke Clinical Research Institute's innovation team. I'm building it alongside my work as a digital product consultant at Method.
What I do think about is the longer arc. The platform already tracks real signal: which documents and guides participants are actually reading, what they're searching for and whether the documents answer it, whether caregivers are engaged or absent. If a participant's search terms suggest an adverse event, that surfaces immediately for their coordinator. If their searches consistently return gaps in the protocol documents, that's information the study team needs.
All of that flows back — to coordinators, to study teams, to sponsors. The goal is improving the participant experience in the trial that's running. But we're already seeing where it points upstream: to where consent documents fail people before a trial even starts, and where better study design might prevent dropout before it becomes a problem.
That's the gap Brian noticed when his father was in that trial. Everyone else had tools. The participant had a stack of paper. It has real consequences — for data quality, for retention, and for the people whose health outcomes depend on staying engaged with their protocol.
Whether we've built exactly the right solution is still being tested. But it feels like a problem worth working on.
Learn more at getcleartrials.com or follow us on LinkedIn.





Member discussion